镧系元素(Ln)作为稀土元素的重要组成部分,是我国重要的战略资源。近年来,探究镧系元素在新能源领域的应用已成为了稀土材料研究的战略方向。随着新能源产业快速发展,锂离子电池的性能需求持续上升,而优化正极材料是改善其性能的关键途径。目前,LiNiO2(LNO)因其高能量密度和低成本优势备受关注,但其化学稳定性、结构稳定性和热稳定性等问题仍有待突破。
对LNO正极材料进行掺杂是有效的优化改性方法。镧系元素具有作为LNO优秀掺杂改性剂的三大特性:1)较大的离子半径;2)与氧的强键合能力;3)电化学惰性。然而,镧系元素掺杂LNO面临双重挑战:实验层面,大离子尺寸镧系元素难以掺入层状材料;模拟计算层面,含镧系元素的体系存在计算成本高、计算收敛性差及结果不稳定等问题。
近日,郑家新团队通过大量第一性原理计算模拟发现,Ln的4f电子在高自旋状态下的多样排布和强关联效应会导致LNO+Ln体系出现多重亚稳态,这正是造成计算难以收敛和结果失准的关键原因。基于此,团队创新开发了基态搜索方法,实现了对LNO+Ln(La-Gd)体系的快速、稳定、准确计算。相关成果以“Ground-State Search and Modification Effects of Lanthanide Substitution in LiNiO2: A First-Principles Study”为题发表于《Science China Materials》(中科院一区Top)。
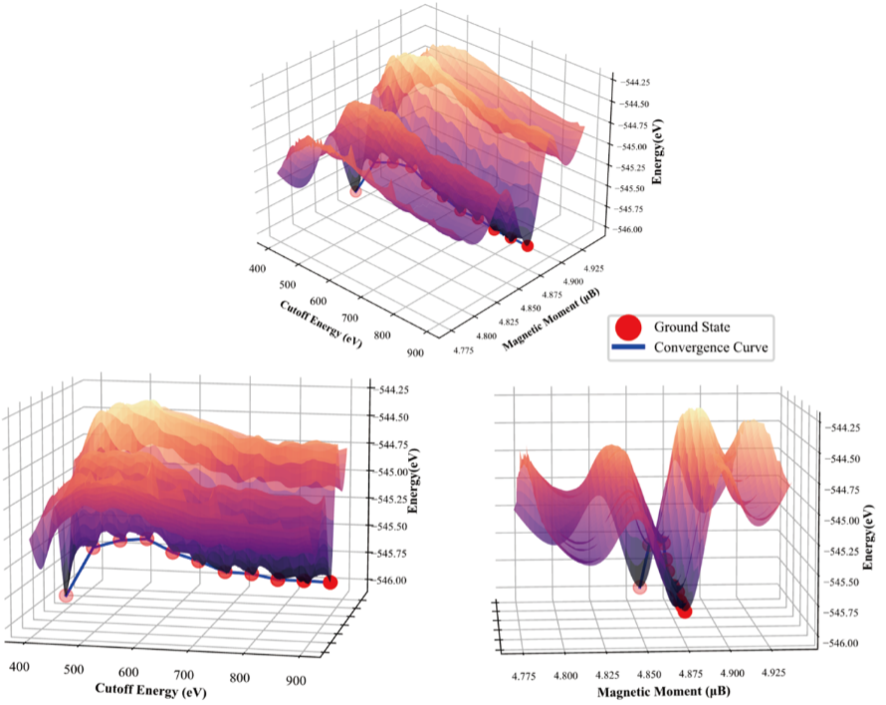
图1. 镧系元素理论计算中的亚稳态和基态概念图
在理论计算中,模型基态电子结构的计算准确性和计算稳定性都与其磁性状态密切相关。在常规理论计算模型中,体系的磁性状态通常可分为高自旋态和低自旋态。这两种状态在能量和磁矩值上存在显著差异,因而在计算收敛时能够被明确区分。郑家新团队研究发现,在LNO+Ln体系中,高自旋态能量更低。特别值得注意的是,镧系元素的计算结果显示,即使磁矩值在小数位级别的微小差距也可能引发约1 eV的能量变化。这种现象源于LNO+Ln基态电子结构附近存在大量亚稳态——这些亚稳态的4f电子磁矩值与基态差别极小,但对应了4f电子的不同排布方式,从而导致能量显著不同。这正是第一性原理计算易收敛于亚稳态,造成收敛性差和结果失准的根本原因。
针对这一稀土元素特有的理论计算难题,郑家新团队创新性提出一种基态搜索方法:通过在计算流程中调整初始磁性收敛控制参数,在DFT+U方法下进行低成本、遍历式的自洽场(SCF)运算搜索。研究发现,收敛态能量与4f电子磁矩值存在明确对应关系,因此可通过磁矩值精准识别电子结构基态。该方法相较常规计算具有三大优势:1)显著降低算力消耗;2)大幅提升计算稳定性与效率;3)确保结果高精度。图2展示了该方法的计算流程和测试表现。
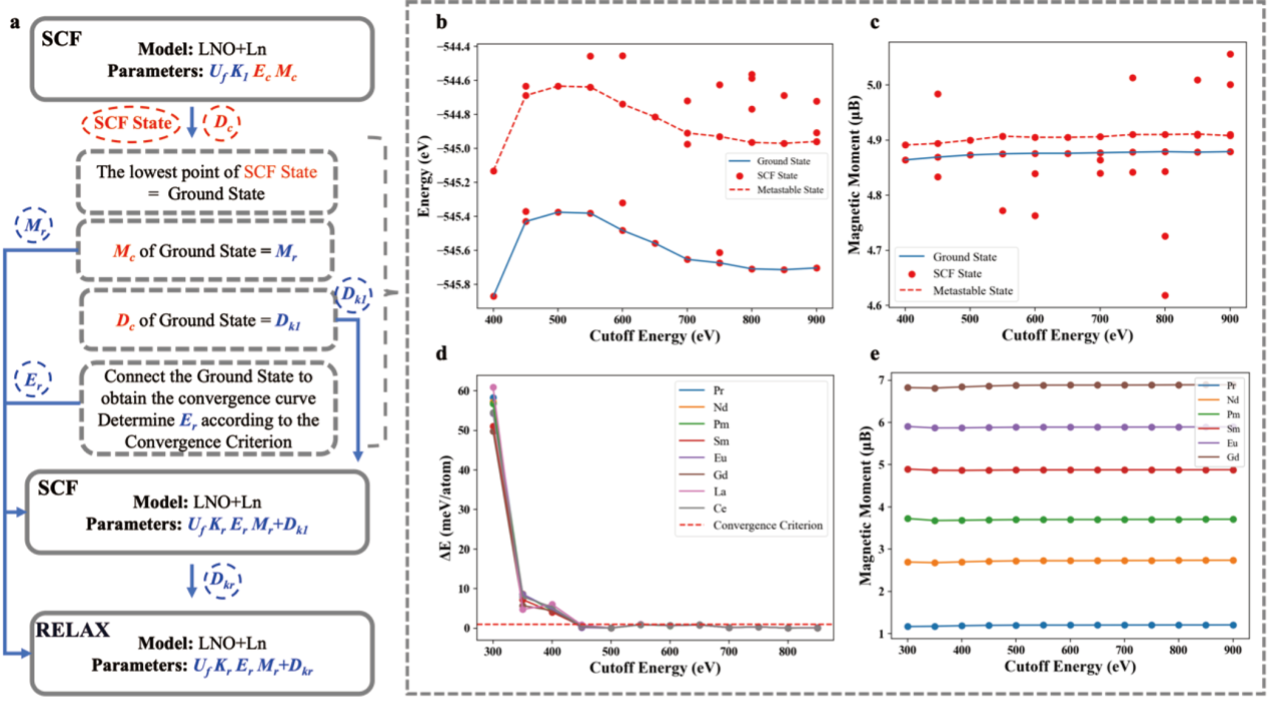
图2. LNO+Ln(La-Gd)的基态搜索方法流程与测试表现
基于基态搜索方法,团队高效获得了LNO+Ln(La-Gd)的精确基态结构(包括晶体结构和电子结构),并进一步计算出Li/Ni反位能和氧空位形成能。研究发现,LNO+Ln掺杂可显著提升LNO的分层有序性、有效抑制Li/Ni反位并增强氧稳定性,这些结论均与近期发表的系列LNO高镍相关实验工作相符合,证实了该方法的准确性和可靠性。在机理分析方面,团队揭示Ln掺杂主要通过以下途径发挥作用:尺寸效应、f电子强关联作用及氧化性氛围共同抑制Li/Ni反位;通过Ln-O键长、键强及电荷转移稳定晶格氧。最终研究表明,在镧系元素中,Ce作为掺杂剂表现出最优异的性能,成为最具应用潜力的候选材料。相关计算结果如图3所示。
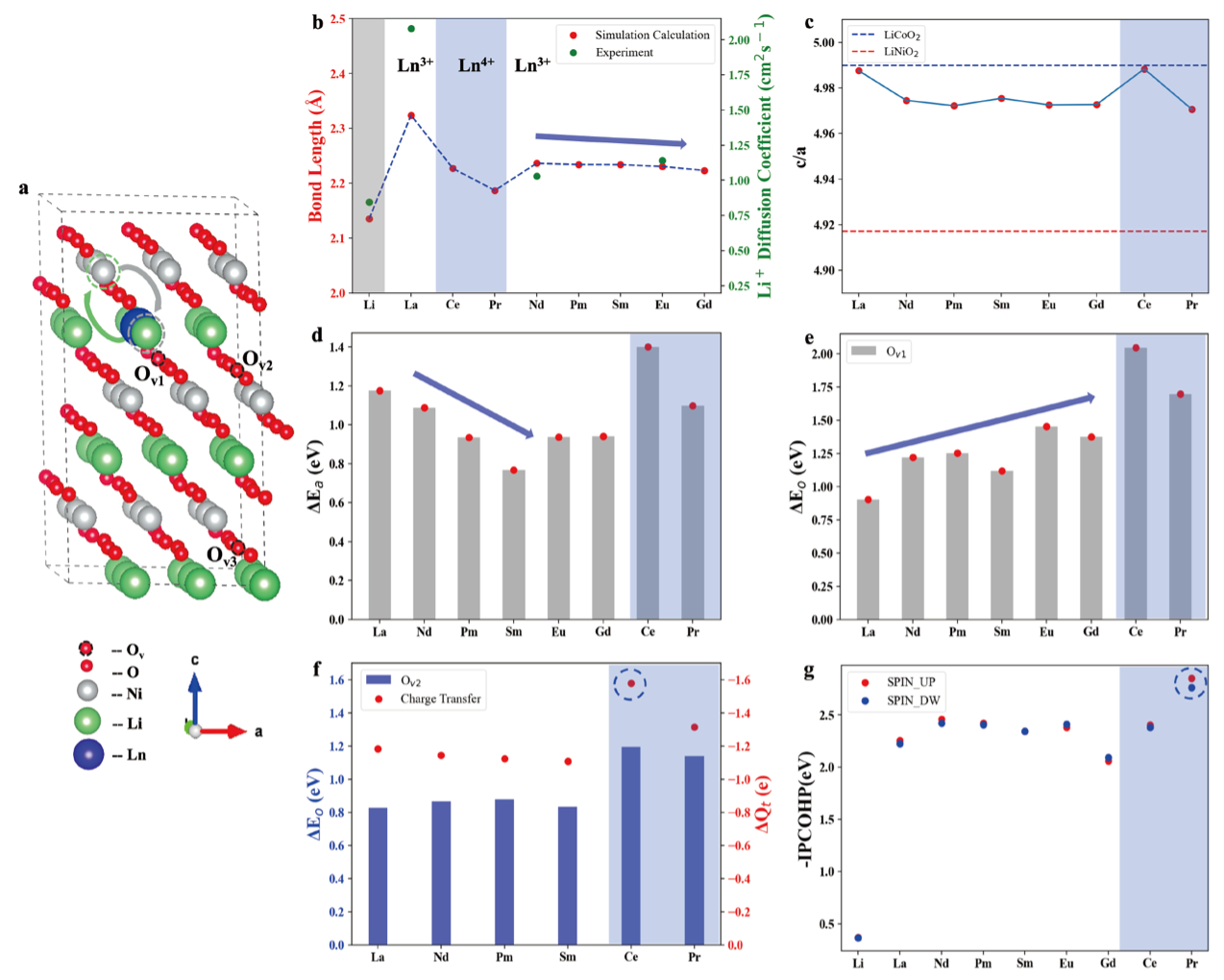
图3. LNO+Ln(La-Gd)的改性研究计算结果
金沙赌场
郑家新副教授与博士后叶耀坤为本文共同通讯作者,2022级硕士研究生吴光银为第一作者。研究得到了国家自然科学基金和北大深研院-屹艮科技电池材料仿真联合实验室的支持。
论文链接://engine.scichina.com/doi/10.1007/s40843-025-3388-0




